Next-generation data filtering in the genomics era
Genomic data are ubiquitous across disciplines, from agriculture to biodiversity, ecology, evolution and human health. However, these datasets often contain noise or errors and are missing information that can affect the accuracy and reliability of subsequent computational analyses and conclusions. A key step in genomic data analysis is filtering — removing sequencing bases, reads, genetic variants and/or individuals from a dataset — to improve data quality for downstream analyses. Researchers are confronted with a multitude of choices when filtering genomic data; they must choose which filters to apply and select appropriate thresholds. To help usher in the next generation of genomic data filtering, we review and suggest best practices to improve the implementation, reproducibility and reporting standards for filter types and thresholds commonly applied to genomic datasets. We focus mainly on filters for minor allele frequency, missing data per individual or per locus, linkage disequilibrium and Hardy–Weinberg deviations. Using simulated and empirical datasets, we illustrate the large effects of different filtering thresholds on common population genetics statistics, such as Tajima’s D value, population differentiation (FST), nucleotide diversity (π) and effective population size (Ne).
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
206,07 € per year
only 17,17 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
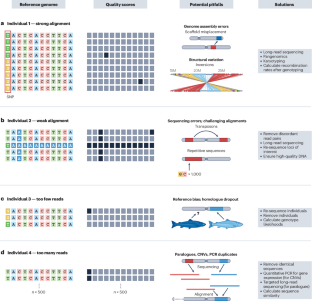
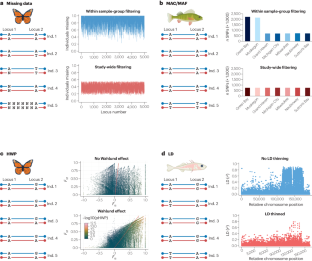
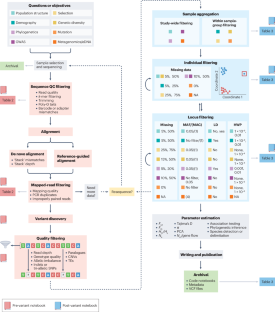
Similar content being viewed by others

Imputation of low-coverage sequencing data from 150,119 UK Biobank genomes
Article Open access 29 June 2023

Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank
Article Open access 29 June 2023

The sequences of 150,119 genomes in the UK Biobank
Article Open access 20 July 2022
Data availability
Information on the empirical and simulated data used for the analyses shown in this review is available in the Supplementary Information.
Code availability
References
- Allendorf, F. W., Hohenlohe, P. A. & Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet.11, 697–709 (2010). ArticleCASPubMedGoogle Scholar
- Athanasopoulou, K., Boti, M. A., Adamopoulos, P. G., Skourou, P. C. & Scorilas, A. Third-generation sequencing: the spearhead towards the radical transformation of modern genomics. Life12, 30 (2022). ArticleCASGoogle Scholar
- Fiedler, P. L. et al. Seizing the moment: the opportunity and relevance of the California Conservation Genomics Project to state and federal conservation policy. J. Hered.113, 589–596 (2022). ArticleCASPubMedPubMed CentralGoogle Scholar
- Hu, T., Chitnis, N., Monos, D. & Dinh, A. Next-generation sequencing technologies: an overview. Hum. Immunol.82, 801–811 (2021). ArticleCASPubMedGoogle Scholar
- Pompanon, F., Bonin, A., Bellemain, E. & Taberlet, P. Genotyping errors: causes, consequences and solutions. Nat. Rev. Genet.6, 847–859 (2005). This review summarizes the sources of many common types of sequencing errors and provides some laboratory and bioinformatic ways to mitigate them.ArticleCASPubMedGoogle Scholar
- Stoler, N. & Nekrutenko, A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform.3, lqab019 (2021). ArticlePubMedPubMed CentralGoogle Scholar
- Fountain, E. D., Pauli, J. N., Reid, B. N., Palsbøll, P. J. & Peery, M. Z. Finding the right coverage: the impact of coverage and sequence quality on single nucleotide polymorphism genotyping error rates. Mol. Ecol. Resour.16, 966–978 (2016). ArticleCASPubMedGoogle Scholar
- O’Leary, S. J., Puritz, J. B., Willis, S. C., Hollenbeck, C. M. & Portnoy, D. S. These aren’t the loci you’re looking for: principles of effective SNP filtering for molecular ecologists. Mol. Ecol.27, 3193–3206 (2018). This helpful review discusses the effects of missing data, MAC and other filters on genotyping error rates for RADseq data.ArticleGoogle Scholar
- Rochette, N. C., Rivera-Colón, A. G. & Catchen, J. M. Stacks 2: analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol.28, 4737–4754 (2019). ArticleCASPubMedGoogle Scholar
- Ahrens, C. W. et al. Regarding the F-word: the effects of data filtering on inferred genotype–environment associations. Mol. Ecol. Resour.21, 1460–1474 (2021). ArticlePubMedGoogle Scholar
- Andrews, K. R. & Luikart, G. Recent novel approaches for population genomics data analysis. Mol. Ecol.23, 1661–1667 (2014). ArticlePubMedGoogle Scholar
- Shafer, A. B. A. et al. Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol. Evol.8, 907–917 (2017). This study demonstrates the effects of different filtering and alignment choices on several downstream statistics and demographic reconstruction in RADseq data.ArticleGoogle Scholar
- Larson, W. A., Isermann, D. A. & Feiner, Z. S. Incomplete bioinformatic filtering and inadequate age and growth analysis lead to an incorrect inference of harvested-induced changes. Evol. Appl.14, 278–289 (2021). ArticleCASPubMedGoogle Scholar
- Nazareno, A. G. & Knowles, L. L. There is no ‘rule of thumb’: genomic filter settings for a small plant population to obtain unbiased gene flow estimates. Front. Plant Sci.12, 677009 (2021). This comprehensive analysis of empirical data demonstrates how missing data and MAF thresholds affect estimates of gene flow.ArticlePubMedPubMed CentralGoogle Scholar
- Sethuraman, A. et al. Continued misuse of multiple testing correction methods in population genetics — a wake-up call? Mol. Ecol. Resour.19, 23–26 (2019). ArticlePubMedGoogle Scholar
- Allendorf, F. W. et al. Conservation and the Genomics of Populations (Oxford Univ. Press, 2022).
- Gervais, L. et al. RAD-sequencing for estimating genomic relatedness matrix-based heritability in the wild: a case study in roe deer. Mol. Ecol. Resour.19, 1205–1217 (2019). ArticleCASPubMedGoogle Scholar
- Crow, J. F. & Kimura, M. An Introduction to Population Genetics Theory (Scientific Publishers, 2017).
- Van Etten, J., Stephens, T. G. & Bhattacharya, D. A k-mer-based approach for phylogenetic classification of taxa in environmental genomic data. Syst. Biol. 72, 1101–1118 (2023). ArticleCASPubMedGoogle Scholar
- Todd, E. V., Black, M. A. & Gemmell, N. J. The power and promise of RNA-seq in ecology and evolution. Mol. Ecol.25, 1224–1241 (2016). ArticleCASPubMedGoogle Scholar
- Conesa, A. et al. A survey of best practices for RNA-seq data analysis. Genome Biol.17, 13 (2016). ArticlePubMedPubMed CentralGoogle Scholar
- Olofsson, D., Preußner, M., Kowar, A., Heyd, F. & Neumann, A. One pipeline to predict them all? On the prediction of alternative splicing from RNA-seq data. Biochem. Biophys. Res. Commun.653, 31–37 (2023). ArticleCASPubMedGoogle Scholar
- Upton, R. N. et al. Design, execution, and interpretation of plant RNA-seq analyses. Front. Plant Sci.14, 1135455 (2023). ArticlePubMedPubMed CentralGoogle Scholar
- Rehn, J. et al. RaScALL: rapid (Ra) screening (Sc) of RNA-seq data for prognostically significant genomic alterations in acute lymphoblastic leukaemia (ALL). PLOS Genet.18, e1010300 (2022). ArticleCASPubMedPubMed CentralGoogle Scholar
- Boshuizen, H. C. & te Beest, D. E. Pitfalls in the statistical analysis of microbiome amplicon sequencing data. Mol. Ecol. Resour.23, 539–548 (2023). ArticlePubMedGoogle Scholar
- Combrink, L. et al. Best practice for wildlife gut microbiome research: a comprehensive review of methodology for 16S rRNA gene investigations. Front. Microbiol.14, 1092216 (2023). ArticlePubMedPubMed CentralGoogle Scholar
- Cheng, Z. et al. Transcriptomic analysis of circulating leukocytes obtained during the recovery from clinical mastitis caused by Escherichia coli in Holstein dairy cows. Animals12, 2146 (2022). ArticlePubMedPubMed CentralGoogle Scholar
- Yang, L. & Chen, J. Benchmarking differential abundance analysis methods for correlated microbiome sequencing data. Brief. Bioinformatics24, bbac607 (2023). ArticlePubMedGoogle Scholar
- Patin, N. V. & Goodwin, K. D. Capturing marine microbiomes and environmental DNA: a field sampling guide. Front. Microbiol.13, 1026596 (2023). ArticlePubMedPubMed CentralGoogle Scholar
- Ruppert, K. M., Kline, R. J. & Rahman, M. S. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: a systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv.17, e00547 (2019). Google Scholar
- Deyneko, I. V. et al. Modeling and cleaning RNA-seq data significantly improve detection of differentially expressed genes. BMC Bioinformatics23, 488 (2022). ArticleCASPubMedPubMed CentralGoogle Scholar
- Giusti, A., Malloggi, C., Magagna, G., Filipello, V. & Armani, A. Is the metabarcoding ripe enough to be applied to the authentication of foodstuff of animal origin? A systematic review. Compr. Rev. Food Sci. Food Saf.23, 1–21 (2024). ArticleGoogle Scholar
- da Fonseca, R. R. et al. Next-generation biology: sequencing and data analysis approaches for non-model organisms. Mar. Genomics30, 3–13 (2016). ArticlePubMedGoogle Scholar
- Zhao, M. et al. Exploring conflicts in whole genome phylogenetics: a case study within manakins (Aves: Pipridae). Syst. Biol.72, 161–178 (2023). ArticleCASPubMedGoogle Scholar
- Koboldt, D. C. Best practices for variant calling in clinical sequencing. Genome Med12, 91 (2020). ArticlePubMedPubMed CentralGoogle Scholar
- Giani, A. M., Gallo, G. R., Gianfranceschi, L. & Formenti, G. Long walk to genomics: history and current approaches to genome sequencing and assembly. Comput. Struct. Biotechnol. J.18, 9–19 (2020). ArticleCASPubMedGoogle Scholar
- Kumar, K. R., Cowley, M. J. & Davis, R. L. Next-generation sequencing and emerging technologies. Semin. Thromb. Hemost.45, 661–673 (2019). ArticleCASPubMedGoogle Scholar
- Shendure, J. et al. DNA sequencing at 40: past, present and future. Nature550, 345–353 (2017). ArticleCASPubMedGoogle Scholar
- Lou, R. N., Jacobs, A., Wilder, A. P. & Therkildsen, N. O. A beginner’s guide to low-coverage whole genome sequencing for population genomics. Mol. Ecol.30, 5966–5993 (2021). This reviews discusses the production and analysis of low-coverage WGS data.ArticlePubMedGoogle Scholar
- Olson, N. D. et al. Variant calling and benchmarking in an era of complete human genome sequences. Nat. Rev. Genet.24, 464–483 (2023). ArticleCASPubMedGoogle Scholar
- Rochette, N. C. & Catchen, J. M. Deriving genotypes from RAD-seq short-read data using Stacks. Nat. Protoc.12, 2640–2659 (2017). ArticleCASPubMedGoogle Scholar
- Paris, J. R., Stevens, J. R. & Catchen, J. M. Lost in parameter space: a road map for stacks. Methods Ecol. Evol.8, 1360–1373 (2017). ArticleGoogle Scholar
- Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M. & Wilson, J. F. Runs of homozygosity: windows into population history and trait architecture. Nat. Rev. Genet.19, 220–234 (2018). ArticleCASPubMedGoogle Scholar
- Heller, R. et al. A reference-free approach to analyse RADseq data using standard next generation sequencing toolkits. Mol. Ecol. Resour.21, 1085–1097 (2021). ArticleCASPubMedGoogle Scholar
- Bohling, J. Evaluating the effect of reference genome divergence on the analysis of empirical RADseq datasets. Ecol. Evol.10, 7585–7601 (2020). ArticlePubMedPubMed CentralGoogle Scholar
- Valiente-Mullor, C. et al. One is not enough: on the effects of reference genome for the mapping and subsequent analyses of short-reads. PLOS Comput. Biol.17, e1008678 (2021). ArticleCASPubMedPubMed CentralGoogle Scholar
- Hendricks, S. et al. Recent advances in conservation and population genomics data analysis. Evol. Appl.11, 1197–1211 (2018). ArticlePubMed CentralGoogle Scholar
- Vaux, F., Dutoit, L., Fraser, C. I. & Waters, J. M. Genotyping-by-sequencing for biogeography. J. Biogeogr.50, 262–281 (2023). ArticleGoogle Scholar
- Jackson, B. C., Campos, J. L. & Zeng, K. The effects of purifying selection on patterns of genetic differentiation between Drosophila melanogaster populations. Heredity114, 163–174 (2015). ArticleCASPubMedGoogle Scholar
- Luikart, G., England, P. R., Tallmon, D., Jordan, S. & Taberlet, P. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet.4, 981–994 (2003). ArticleCASPubMedGoogle Scholar
- Benestan, L. et al. Sex matters in massive parallel sequencing: evidence for biases in genetic parameter estimation and investigation of sex determination systems. Mol. Ecol.26, 6767–6783 (2017). ArticleCASPubMedGoogle Scholar
- Yang, Z. et al. Multi-omics provides new insights into the domestication and improvement of dark jute (Corchorus olitorius). Plant J.112, 812–829 (2022). ArticleCASPubMedGoogle Scholar
- Zeng, L. et al. Whole genomes and transcriptomes reveal adaptation and domestication of pistachio. Genome Biol.20, 79 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Zhernakova, D. V. et al. Genome-wide sequence analyses of ethnic populations across Russia. Genomics112, 442–458 (2020). ArticleCASPubMedGoogle Scholar
- Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods9, 357–359 (2012). ArticleCASPubMedPubMed CentralGoogle Scholar
- Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics25, 1754–1760 (2009). ArticleCASPubMedPubMed CentralGoogle Scholar
- Pfeifer, S. P. From next-generation resequencing reads to a high-quality variant data set. Heredity118, 111–124 (2017). ArticleCASPubMedGoogle Scholar
- Lefouili, M. & Nam, K. The evaluation of BCFtools mpileup and GATK HaplotypeCaller for variant calling in non-human species. Sci. Rep.12, 11331 (2022). ArticleCASPubMedPubMed CentralGoogle Scholar
- Chen, N.-C., Solomon, B., Mun, T., Iyer, S. & Langmead, B. Reference flow: reducing reference bias using multiple population genomes. Genome Biol.22, 8 (2021). ArticlePubMedPubMed CentralGoogle Scholar
- Günther, T. & Nettelblad, C. The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLOS Genet.15, e1008302 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Rhie, A. et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature592, 737–746 (2021). ArticleCASPubMedPubMed CentralGoogle Scholar
- Ho, S. S., Urban, A. E. & Mills, R. E. Structural variation in the sequencing era. Nat. Rev. Genet.21, 171–189 (2020). ArticleCASPubMedGoogle Scholar
- Singh, A. K. et al. Detecting copy number variation in next generation sequencing data from diagnostic gene panels. BMC Med. Genomics14, 214 (2021). ArticleCASPubMedPubMed CentralGoogle Scholar
- Willis, S. C., Hollenbeck, C. M., Puritz, J. B., Gold, J. R. & Portnoy, D. S. Haplotyping RAD loci: an efficient method to filter paralogs and account for physical linkage. Mol. Ecol. Resour.17, 955–965 (2017). ArticleCASPubMedGoogle Scholar
- Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol.20, 275 (2019). ArticleCASPubMedPubMed CentralGoogle Scholar
- Rochette, N. C. et al. On the causes, consequences, and avoidance of PCR duplicates: towards a theory of library complexity. Mol. Ecol. Resour.23, 1299–1318 (2023). ArticleCASPubMedGoogle Scholar
- Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, 2020).
- Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics15, 356 (2014). ArticlePubMedPubMed CentralGoogle Scholar
- Eaton, D. A. R. & Overcast, I. ipyrad: interactive assembly and analysis of RADseq datasets. Bioinformatics36, 2592–2594 (2020). ArticleCASPubMedGoogle Scholar
- Layer, R. M., Chiang, C., Quinlan, A. R. & Hall, I. M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol.15, R84 (2014). ArticlePubMedPubMed CentralGoogle Scholar
- Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience10, giab008 (2021). ArticlePubMedPubMed CentralGoogle Scholar
- Mona, S., Benazzo, A., Delrieu-Trottin, E. & Lesturgie, P. Population genetics using low coverage RADseq data in non-model organisms: biases and solutions. Preprint at Authoreahttps://doi.org/10.22541/au.168252801.19878064/v1 (2023).
- Nielsen, R., Korneliussen, T., Albrechtsen, A., Li, Y. & Wang, J. SNP calling, genotype calling, and sample allele frequency estimation from new-generation sequencing data. PLoS ONE7, e37558 (2012). ArticleCASPubMedPubMed CentralGoogle Scholar
- Warmuth, V. M. & Ellegren, H. Genotype-free estimation of allele frequencies reduces bias and improves demographic inference from RADseq data. Mol. Ecol. Resour.19, 586–596 (2019). ArticleCASPubMedGoogle Scholar
- Wright, B. et al. From reference genomes to population genomics: comparing three reference-aligned reduced-representation sequencing pipelines in two wildlife species. BMC Genomics20, 453 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Huang, H. & Knowles, L. L. Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Syst. Biol.65, 357–365 (2016). ArticleCASPubMedGoogle Scholar
- Duntsch, L., Whibley, A., Brekke, P., Ewen, J. G. & Santure, A. W. Genomic data of different resolutions reveal consistent inbreeding estimates but contrasting homozygosity landscapes for the threatened Aotearoa New Zealand hihi. Mol. Ecol.30, 6006–6020 (2021). ArticleCASPubMedGoogle Scholar
- Kardos, M. & Waples, R. S. Low-coverage sequencing and Wahlund effect severely bias estimates of inbreeding, heterozygosity, and effective population size in North American wolves. Mol. Ecol. https://doi.org/10.1111/mec.17415 (2024). This study reports biases that could affect management decisions caused by next-generation sequencing filtering choices, low-coverage data and the sampling strategy.
- Schmidt, T. L., Jasper, M.-E., Weeks, A. R. & Hoffmann, A. A. Unbiased population heterozygosity estimates from genome-wide sequence data. Methods Ecol. Evol.12, 1888–1898 (2021). ArticleGoogle Scholar
- Sopniewski, J. & Catullo, R. A. Estimates of heterozygosity from single nucleotide polymorphism markers are context-dependent and often wrong. Mol. Ecol. Resour.24, e13947 (2024). ArticleCASPubMedGoogle Scholar
- Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics155, 945–959 (2000). ArticleCASPubMedPubMed CentralGoogle Scholar
- Waples, R. S. Testing for Hardy–Weinberg proportions: have we lost the plot? J. Hered.106, 1–19 (2015). ArticlePubMedGoogle Scholar
- Gautier, M. et al. The effect of RAD allele dropout on the estimation of genetic variation within and between populations. Mol. Ecol.22, 3165–3178 (2013). ArticleCASPubMedGoogle Scholar
- McKinney, G. J., Waples, R. K., Seeb, L. W. & Seeb, J. E. Paralogs are revealed by proportion of heterozygotes and deviations in read ratios in genotyping-by-sequencing data from natural populations. Mol. Ecol. Resour.17, 656–669 (2017). ArticleCASPubMedGoogle Scholar
- Bitarello, B. D., Brandt, D. Y. C., Meyer, D. & Andrés, A. M. Inferring balancing selection from genome-scale data. Genome Biol. Evol.15, evad032 (2023). ArticlePubMedPubMed CentralGoogle Scholar
- Pearman, W. S., Urban, L. & Alexander, A. Commonly used Hardy–Weinberg equilibrium filtering schemes impact population structure inferences using RADseq data. Mol. Ecol. Resour.22, 2599–2613 (2022). This study demonstrates the impact of pooling or splitting sample-groups when applying HWP filters toFSTand other population structure inferences.ArticleCASPubMedPubMed CentralGoogle Scholar
- Linderoth, T. P. Identifying population histories, adaptive genes, and genetic duplication from population-scale next generation sequencing. Genome Res.20, 291–300 (2018). Google Scholar
- Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol.57, 289–300 (1995). ArticleGoogle Scholar
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat.6, 65–70 (1979). Google Scholar
- Graffelman, J., Jain, D. & Weir, B. A genome-wide study of Hardy–Weinberg equilibrium with next generation sequence data. Hum. Genet.136, 727–741 (2017). ArticleCASPubMedPubMed CentralGoogle Scholar
- Larson, W. A. et al. Genotyping by sequencing resolves shallow population structure to inform conservation of Chinook salmon (Oncorhynchus tshawytscha). Evol. Appl.7, 355–369 (2014). ArticleCASPubMedPubMed CentralGoogle Scholar
- Waples, R. K., Larson, W. A. & Waples, R. S. Estimating contemporary effective population size in non-model species using linkage disequilibrium across thousands of loci. Heredity117, 233–240 (2016). ArticleCASPubMedPubMed CentralGoogle Scholar
- Gattepaille, L. M., Jakobsson, M. & Blum, M. G. Inferring population size changes with sequence and SNP data: lessons from human bottlenecks. Heredity110, 409–419 (2013). ArticleCASPubMedPubMed CentralGoogle Scholar
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics123, 585 LP–585595 (1989). ArticleGoogle Scholar
- Arantes, L. S. et al. Scaling-up RADseq methods for large datasets of non-invasive samples: lessons for library construction and data preprocessing. Mol. Ecol. Resour. https://doi.org/10.1111/1755-0998.13859 (2023).
- Cubry, P., Vigouroux, Y. & François, O. The empirical distribution of singletons for geographic samples of DNA sequences. Front. Genet.8, 139 (2017). ArticlePubMedPubMed CentralGoogle Scholar
- Linck, E. & Battey, C. J. Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Mol. Ecol. Resour.19, 639–647 (2019). This study demonstrates how MAF thresholds affect population structure inferences using both simulated and empirical data.ArticleCASPubMedGoogle Scholar
- Andersson, B. A., Zhao, W., Haller, B. C., Brännström, Å. & Wang, X.-R. Inference of the distribution of fitness effects of mutations is affected by single nucleotide polymorphism filtering methods, sample size and population structure. Mol. Ecol. Resour.23, 1589–1603 (2023). ArticleCASPubMedGoogle Scholar
- Díaz-Arce, N. & Rodríguez-Ezpeleta, N. Selecting RAD-seq data analysis parameters for population genetics: the more the better? Front. Genet.10, 533 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Holsinger, K. E. & Weir, B. S. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat. Rev. Genet.10, 639–650 (2009). ArticleCASPubMedPubMed CentralGoogle Scholar
- Roesti, M., Salzburger, W. & Berner, D. Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evol. Biol.12, 94 (2012). ArticlePubMedPubMed CentralGoogle Scholar
- Yin, X. et al. Rapid, simultaneous increases in the effective sizes of adaptively divergent yellow perch (Perca flavescens) populations. Preprint at bioRxivhttps://doi.org/10.1101/2024.04.21.590447 (2024).
- Visscher, P. M. et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet.101, 5–22 (2017). ArticleCASPubMedPubMed CentralGoogle Scholar
- Tennessen, J. A. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science337, 64–69 (2012). ArticleCASPubMedPubMed CentralGoogle Scholar
- Dementieva, N. V. et al. Assessing the effects of rare alleles and linkage disequilibrium on estimates of genetic diversity in the chicken populations. Animal15, 100171 (2021). ArticleCASPubMedGoogle Scholar
- De Meeûs, T. Revisiting FIS, FST, Wahlund effects, and null alleles. J. Hered.109, 446–456 (2018). ArticlePubMedGoogle Scholar
- Levy-Sakin, M. et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun.10, 1025 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Zhang, H., Yin, L., Wang, M., Yuan, X. & Liu, X. Factors affecting the accuracy of genomic selection for agricultural economic traits in maize, cattle, and pig populations. Front. Genet.10, 189 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Anderson, E. C. & Garza, J. C. The power of single-nucleotide polymorphisms for large-scale parentage inference. Genetics172, 2567–2582 (2006). ArticleCASPubMedPubMed CentralGoogle Scholar
- Dussault, F. M. & Boulding, E. G. Effect of minor allele frequency on the number of single nucleotide polymorphisms needed for accurate parentage assignment: a methodology illustrated using Atlantic salmon. Aquac. Res.49, 1368–1372 (2018). ArticleGoogle Scholar
- Thompson, E. The estimation of pairwise relationships. Ann. Hum. Genet.39, 173–188 (1975). ArticleCASPubMedGoogle Scholar
- Goubert, C. et al. A beginner’s guide to manual curation of transposable elements. Mob. DNA13, 7 (2022). ArticlePubMedPubMed CentralGoogle Scholar
- Storer, J. M., Hubley, R., Rosen, J. & Smit, A. F. A. Curation guidelines for de novo generated transposable element families. Curr. Protoc.1, e154 (2021). ArticlePubMedPubMed CentralGoogle Scholar
- Hemstrom, W. B., Freedman, M. G., Zalucki, M. P., Ramírez, S. R. & Miller, M. R. Population genetics of a recent range expansion and subsequent loss of migration in monarch butterflies. Mol. Ecol.31, 4544–4557 (2022). ArticlePubMedPubMed CentralGoogle Scholar
- Escoda, L., González-Esteban, J., Gómez, A. & Castresana, J. Using relatedness networks to infer contemporary dispersal: application to the endangered mammal Galemys pyrenaicus. Mol. Ecol.26, 3343–3357 (2017). ArticlePubMedGoogle Scholar
- Brown, A. V. et al. Ten quick tips for sharing open genomic data. PLOS Comput. Biol.14, e1006472 (2018). ArticlePubMedPubMed CentralGoogle Scholar
- Zhang, D. et al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour.20, 348–355 (2020). ArticlePubMedGoogle Scholar
- Tanjo, T., Kawai, Y., Tokunaga, K., Ogasawara, O. & Nagasaki, M. Practical guide for managing large-scale human genome data in research. J. Hum. Genet.66, 39–52 (2021). ArticlePubMedGoogle Scholar
- Del Fabbro, C., Scalabrin, S., Morgante, M. & Giorgi, F. M. An extensive evaluation of read trimming effects on illumina NGS data analysis. PLoS ONE8, e85024 (2013). ArticlePubMedPubMed CentralGoogle Scholar
- Yang, S.-F., Lu, C.-W., Yao, C.-T. & Hung, C.-M. To trim or not to trim: effects of read trimming on the de novo genome assembly of a widespread East Asian passerine, the rufous-capped babbler (Cyanoderma ruficeps Blyth). Genes10, 737 (2019). ArticleCASPubMedPubMed CentralGoogle Scholar
- Hotaling, S. et al. Demographic modelling reveals a history of divergence with gene flow for a glacially tied stonefly in a changing post-Pleistocene landscape. J. Biogeogr.45, 304–317 (2018). ArticleGoogle Scholar
- Cumer, T. et al. Double-digest RAD-sequencing: do pre- and post-sequencing protocol parameters impact biological results? Mol. Genet. Genomics296, 457–471 (2021). ArticleCASPubMedGoogle Scholar
- Mastretta-Yanes, A. et al. Restriction site-associated DNA sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Mol. Ecol. Resour.15, 28–41 (2015). ArticleCASPubMedGoogle Scholar
- Ebbert, M. T. W. et al. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinformatics17, 239 (2016). ArticlePubMedPubMed CentralGoogle Scholar
- Euclide, P. T. et al. Attack of the PCR clones: rates of clonality have little effect on RAD-seq genotype calls. Mol. Ecol. Resour.20, 66–78 (2020). ArticleCASPubMedGoogle Scholar
- Flanagan, S. P. & Jones, A. G. Substantial differences in bias between single-digest and double-digest RAD-seq libraries: a case study. Mol. Ecol. Resour.18, 264–280 (2018). ArticleCASPubMedGoogle Scholar
- Martins, F. B. et al. A semi-automated SNP-based approach for contaminant identification in biparental polyploid populations of tropical forage grasses. Front. Plant Sci.12, 737919 (2021). ArticlePubMedPubMed CentralGoogle Scholar
- Deo, T. G. et al. High-resolution linkage map with allele dosage allows the identification of regions governing complex traits and apospory in guinea grass (Megathyrsus maximus). Front. Plant Sci.11, 15 (2020). ArticlePubMedPubMed CentralGoogle Scholar
- Zhang, F. et al. Ancestry-agnostic estimation of DNA sample contamination from sequence reads. Genome Res.30, 185–194 (2020). ArticleCASPubMedPubMed CentralGoogle Scholar
- Christie, M. R., Marine, M. L., Fox, S. E., French, R. A. & Blouin, M. S. A single generation of domestication heritably alters the expression of hundreds of genes. Nat. Commun.7, 10676 (2016). ArticleCASPubMedPubMed CentralGoogle Scholar
- Lou, R. N. & Therkildsen, N. O. Batch effects in population genomic studies with low-coverage whole genome sequencing data: causes, detection and mitigation. Mol. Ecol. Resour.22, 1678–1692 (2022). ArticleCASPubMedGoogle Scholar
- Danecek, P. et al. The variant call format and VCFtools. Bioinformatics27, 2156–2158 (2011). ArticleCASPubMedPubMed CentralGoogle Scholar
- Mirchandani, C. D. et al. A fast, reproducible, high-throughput variant calling workflow for population genomics. Mol. Biol. Evol.41, msad270 (2024). ArticlePubMedGoogle Scholar
- Peñalba, J. V., Peters, J. L. & Joseph, L. Sustained plumage divergence despite weak genomic differentiation and broad sympatry in sister species of Australian woodswallows (Artamus spp.). Mol. Ecol.31, 5060–5073 (2022). ArticlePubMedGoogle Scholar
- Thompson, N. F. et al. A complex phenotype in salmon controlled by a simple change in migratory timing. Science370, 609–613 (2020). ArticleCASPubMedGoogle Scholar
- Howe, K. et al. Significantly improving the quality of genome assemblies through curation. Gigascience10, giaa153 (2021). ArticlePubMedPubMed CentralGoogle Scholar
- Nurk, S. et al. The complete sequence of a human genome. Science376, 44–53 (2022). ArticleCASPubMedPubMed CentralGoogle Scholar
- Michael, T. P. & VanBuren, R. Building near-complete plant genomes. Genome Stud. Mol. Genet.54, 26–33 (2020). CASGoogle Scholar
- Tettelin, H. & Medini, D. The Pangenome: Diversity, Dynamics and Evolution of Genomes (Springer, 2020).
- Wang, T. et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature604, 437–446 (2022). ArticleCASPubMedPubMed CentralGoogle Scholar
- Hemstrom, W. Thirty-Four Kilometers and Fifteen Years: Rapid Adaptation at a Novel Chromosomal Inversion in Recently Introduced Deschutes River Three-Spined Stickleback. Thesis, Oregon State Univ. (2016).
- Halvorsen, S., Korslund, L., Mattingsdal, M. & Slettan, A. Estimating number of European eel (Anguilla anguilla) individuals using environmental DNA and haplotype count in small rivers. Ecol. Evol.13, e9785 (2023). ArticlePubMedPubMed CentralGoogle Scholar
- Whitlock, M. C. & Lotterhos, K. E. Reliable detection of loci responsible for local adaptation: inference of a null model through trimming the distribution of FST. Am. Nat.186, S24–S36 (2015). ArticlePubMedGoogle Scholar
- vonHoldt, B. M. et al. Demographic history shapes North American gray wolf genomic diversity and informs species’ conservation. Mol. Ecol.33, e17231 (2024). ArticleCASPubMedGoogle Scholar
- Alonso-Blanco, C. et al. 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell166, 481–491 (2016). ArticleGoogle Scholar
- Maruki, T., Ye, Z. & Lynch, M. Evolutionary genomics of a subdivided species. Mol. Biol. Evol.39, msac152 (2022). ArticleCASPubMedPubMed CentralGoogle Scholar
- Kessler, C., Wootton, E. & Shafer, A. B. A. Speciation without gene-flow in hybridizing deer. Mol. Ecol.32, 1117–1132 (2023). ArticleCASPubMedGoogle Scholar
- Martchenko, D. & Shafer, A. B. A. Contrasting whole-genome and reduced representation sequencing for population demographic and adaptive inference: an alpine mammal case study. Heredity131, 273–281 (2023). ArticleCASPubMedGoogle Scholar
- Lowy-Gallego, E. et al. Variant calling on the GRCh38 assembly with the data from phase three of the 1000 Genomes Project. Wellcome Open Res. 4, 50 (2019). ArticlePubMedPubMed CentralGoogle Scholar
- Schweizer, R. M. et al. Broad concordance in the spatial distribution of adaptive and neutral genetic variation across an elevational gradient in deer mice. Mol. Biol. Evol.38, 4286–4300 (2021). ArticleCASPubMedPubMed CentralGoogle Scholar
- Kardos, M. et al. Inbreeding depression explains killer whale population dynamics. Nat. Ecol. Evol.7, 675–686 (2023). ArticlePubMedGoogle Scholar
- Malison, R. L. et al. Landscape connectivity and genetic structure in a mainstem and a tributary stonefly (Plecoptera) species using a novel reference genome. J. Hered.113, 453–471 (2022). ArticleCASPubMedGoogle Scholar
- Robinson, J. M. et al. Traditional ecological knowledge in restoration ecology: a call to listen deeply, to engage with, and respect Indigenous voices. Restor. Ecol.29, e13381 (2021). ArticleGoogle Scholar
- Lynch, M. The Origins of Genome Architecture (Sinauer Associates, 2007).
- Lynch, M. & O’Hely, M. Captive breeding and the genetic fitness of natural populations. Conserv. Genet.2, 363–378 (2001). ArticleGoogle Scholar
Acknowledgements
The authors thank E. Anderson, A. Leaché, M. Kardos and the reviewers for their helpful comments that greatly improved this manuscript. The authors also thank M. Exposito-Alonso and the 1001 Genomes Consortium, the 1000 Genomes Project, B. Hand, M. Freedman, M. Kardos, C. Kessler, M. Lynch, R. Malison, D. Martchenko, M. Miller, R. Schweizer, A.B.A. Shafer and X. Yin for allowing their datasets to be reviewed and re-filtered. M.R.C. was funded, in part, by NSF DEB-1856710 and OCE-1924505. G.L. was funded, in part, by NSF-DOB-M66230.
Author information
- These authors contributed equally: William Hemstrom, Jared A. Grummer.
Authors and Affiliations
- Department of Biological Sciences, Purdue University, West Lafayette, IN, USA William Hemstrom & Mark R. Christie
- Flathead Lake Biological Station, Wildlife Biology Program and Division of Biological Sciences, University of Montana, Missoula, MT, USA Jared A. Grummer & Gordon Luikart
- Department of Forestry and Natural Resources, Purdue University, West Lafayette, IN, USA Mark R. Christie
- William Hemstrom
